 上手机找货源
上手机找货源
●凡本网注明“来源:仪器批发网”的所有作品,版权均属于仪器批发网,转载请必须注明仪器批发网,http://www.china17pf.com/,违反者本网将追究相关法律责任。
●本网转载并注明自其它来源的作品,目的在于传递更多信息,并不代表本网赞同其观点或证实其内容的真实性,不承担此类作品侵权行为的直接责任及连带责任。其他媒体、网站或个人从本网转载时,必须保留本网注明的作品来源,并自负版权等法律责任。
●如涉及作品内容、版权等问题,请在作品发表之日起一周内与本网联系,否则视为放弃相关权利。
因而,提取完成后应测定 RNA 的浓度及吸光度。通常,在 260,280,230 nm 处分别测定其吸光度(A260、A280、A230)并计算其比值(A260 /A280,A260 /A230)。

有!设计引物的时候用「RNAstructure」预测一下引物的二级结构即可。

1. 打开「RNAstructure」,单击左上角的「new sequence」按钮:

2. 新建序列文件,会出现一个对话框,输入待检测的引物序列:
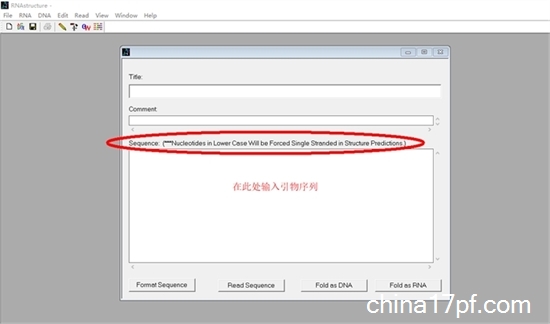
3. 单击「Fold as DNA」,「save changes」,出现新的对话框后直接点击「Start」:
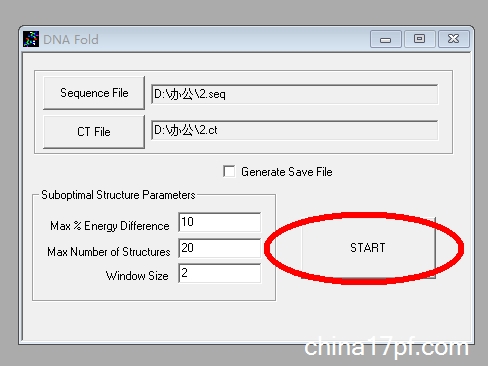
4. 无需更改其他参数,再次点击「Draw structure」,即可得到目标序列的二级结构:
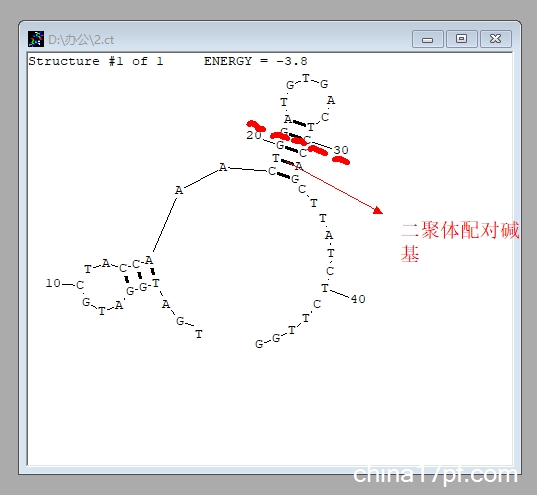
若在特异性扩增的温度范围内不能使 Ct 值差异为 1,则需考虑重新设计引物。


●凡本网注明“来源:仪器批发网”的所有作品,版权均属于仪器批发网,转载请必须注明仪器批发网,http://www.china17pf.com/,违反者本网将追究相关法律责任。
●本网转载并注明自其它来源的作品,目的在于传递更多信息,并不代表本网赞同其观点或证实其内容的真实性,不承担此类作品侵权行为的直接责任及连带责任。其他媒体、网站或个人从本网转载时,必须保留本网注明的作品来源,并自负版权等法律责任。
●如涉及作品内容、版权等问题,请在作品发表之日起一周内与本网联系,否则视为放弃相关权利。

